| Steps | Download files | |
| 1) Inputs Structures of the energy-minimized ligand (red) and apo target (green) molecules in Protein Databank (*.pdb) format |
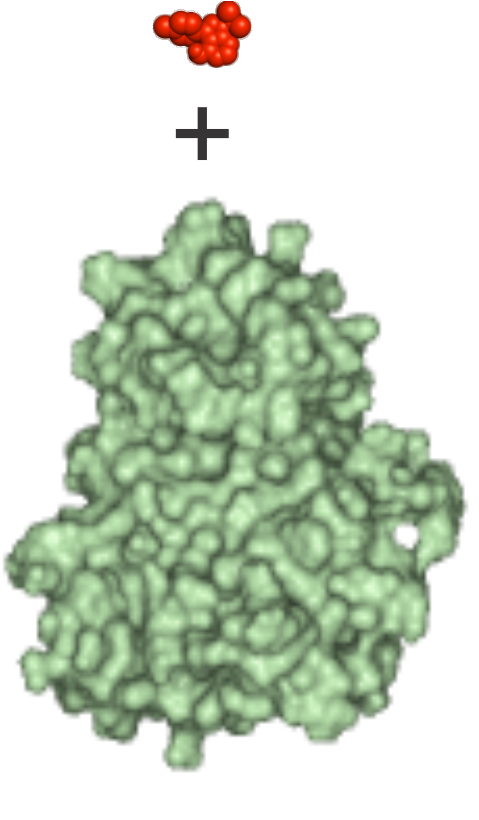 |
Input files 1qcf_ligand.pdb 1qcf_target.pdb |
| 2) Pre-wrapper 2a) Generation of *.pdbqt files with partial charges and atom types. Setting up parameter files for calculation of grid maps (*.gpf) and docking (*.dpf) 2b) The docking box covers the entire target surface (red box, blind docking mode) 2c) Modification of the AD4_parameters.dat file (need to be done only once before the first wrapping cycle) |
 |
Generated files 1qcf_ligand.pdbqt 1qcf_target.pdbqt 1qcf_target.gpf 1qcf_target.dpf Modified AutoDock files AD4_parameters.dat autocomm.h |
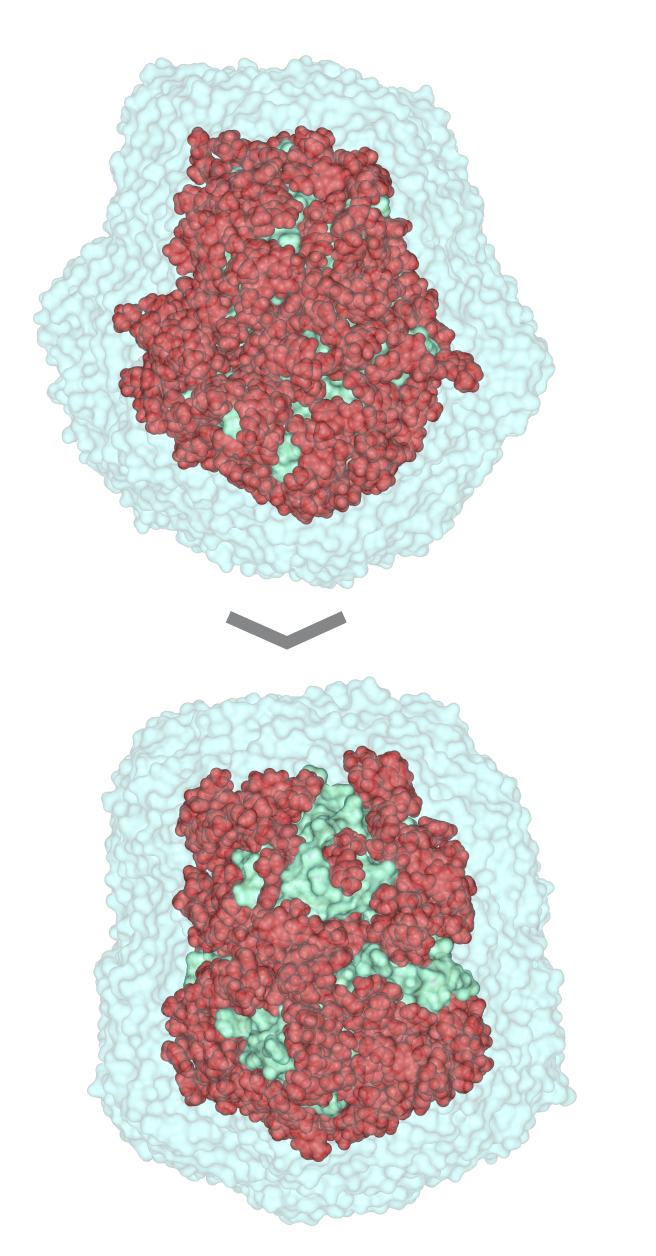
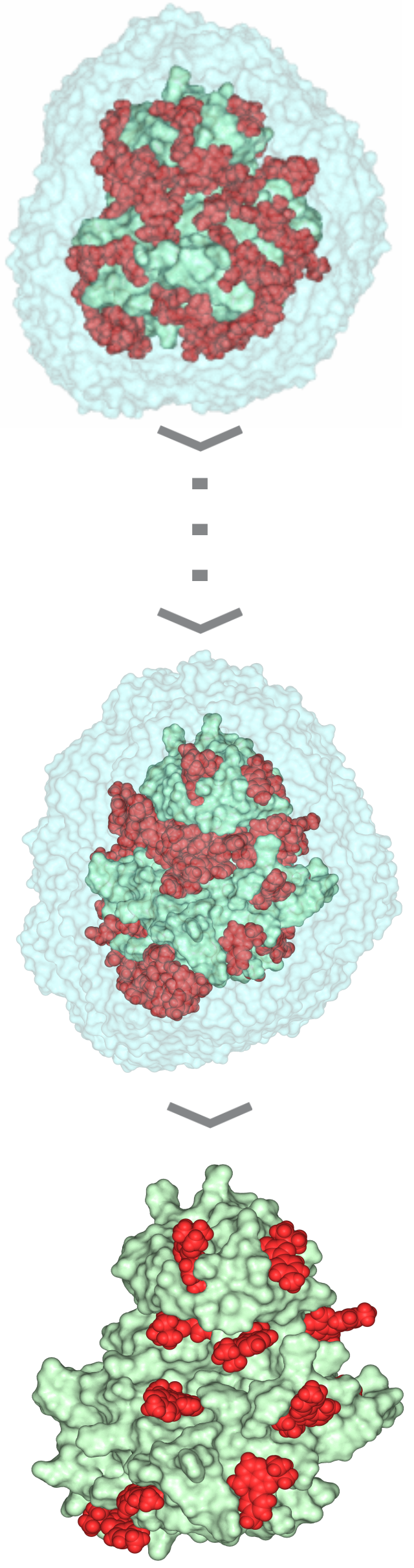
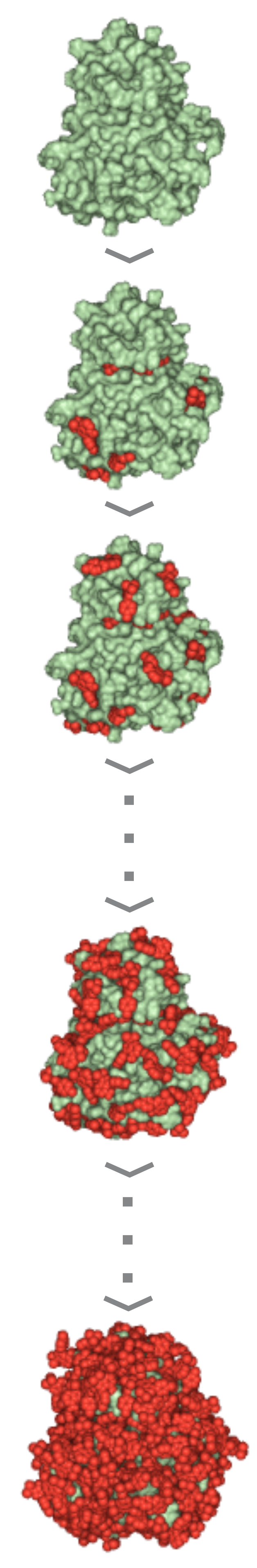
| 3) Wrapper cycle 1 3a) Grid maps (*.map) and additional files (*.glg, *.fld, *.xyz) are produced by Autogrid 4.2 3b) Docking log file (*.dlg) is produced by AutoDock 4.2. 100 blind docking runs per cycle are performed 3c) The *.dlg file is used as input by program Wrp, which first ranks and clusters the docked ligand conformations. The results are reported in a *.sta file 3d) New atom types (YY, LL) of excluded atoms are assigned by wrp. The corresponding *.YY.map and *.LL.map files are generated before the cycle. The resulted *.wrp.pdbqt file contains the docked ligand copies (cluster representatives) 3e) Available free Accessible Surface Area (ASA) is calculated by Msroll 3f) Wrapping ends if ASA ≤ 1 % or the interaction energy value of any cluster representant in the cycle is ≥ 0 kcal/mol. Otherwise, the *.wrp.pdbqt file is forwarded to the next cycle 4) Wrapper cycles 2-16 The process described at Cycle 1 is repeated in consecutive cycles. Wrapping will end if a criterion of Point 3f is met. The complete Wrapping process with all files of the 16 wrapping cycles can be downloaded in a single package (O_1qcf_wrp.tgz) |
 |
Autogrid 4.2 outputs 1qcf_target.YY.map.gz 1qcf_target.LL.map.gz 1qcf_target.glg etc_grid.tgz AutoDock 4.2 output 1qcf_1.dlg Gromacs output (ver. 1.1) O_1qcf_1_ASA_free.log Msroll output (ver. 1.0) O_1qcf_1_ASA_free.log Wrp outputs O_1qcf_1.log O_1qcf_1_input_t.pdb O_1qcf_1_wrp.sta O_1qcf_1_wrp.pdbqt etc_wrp.tgz Wrapper outputs (Cycles 1-16) O_1qcf_wrp.tgz |
| 5) Trimming After the last (16th) wrapping cycle a trimming step is involved to remove ligand copies positioned far from the target surface. The *trm.pdb file contains the target structure wrapped in a monolayer of N=143 ligand copies and can be used in Shaker. |
Input files 1qcf_16_wrp.pdbqt 1qcf_ligand_template.pdb 1qcf_ligand_template.pdbqt Output file O_1qcf_16_wrp.log O_1qcf_16_wrp_trm.pdb |
|